










au sommaire
Les différents types de la maladie de Creutzfeldt-Jakobmaladie de Creutzfeldt-Jakob se différencient par l'âge de développement des symptômessymptômes, par les signes cliniques et par la localisation du prionprion anormal dans les tissus.

Le prion, responsable de la maladie de Creutzfeldt-Jakob. © sbl.salk.edu
Comment peut-on faire la différence entre les différentes formes de la maladie de Creutzfeldt-Jakob puisque la même protéineprotéine PrP anormale est impliquée ? On peut se fier à différents critères.
L'âge des premiers symptômes
Tout d'abord l'âge, puisque les cas sporadiques apparaissent après 60 ans, et les cas génétiquesgénétiques après 40 ans. Si les patients sont jeunes, il y a de fortes chances qu'il s'agisse d'une forme infectieuse. Une étude génétique permet aussi de déterminer la présence de mutations et de vérifier l'hypothèse de la forme génétique. Les antécédents dans la famille des patients permettent aussi de déterminer l'origine de la maladie.
Les signes cliniques
Les signes cliniques de la maladie sont aussi un peu différents entre les trois formes. La maladie de Creutzfeldt-Jakob sporadique évolue en 6 mois, donc très rapidement, et donne une atteinte des fonctions intellectuelles (démencedémence) associée à des myoclonies (des secousses musculaires).
Chez les patients atteints de la variante de la maladie de Creutzfeldt-Jakob (liée à la vache follevache folle), les symptômes commencent par des troubles psychiatriques, des douleursdouleurs, les troubles de l'équilibre et la démence apparaissent ensuite. La durée d'évolution est plus longue, environ 18 mois en moyenne.
Pour la maladie de Creutzfeldt-Jakob liée aux hormoneshormones de croissance, les troubles de l'équilibre arrivent également avant la démence. L'évolution dure aussi en moyenne 18 mois.

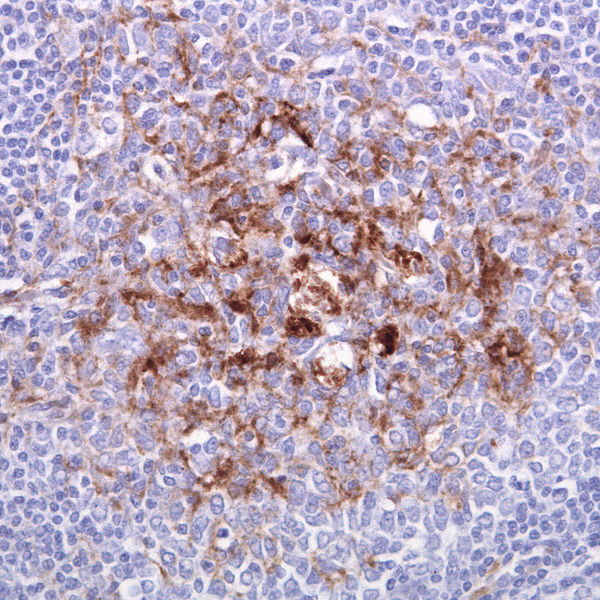
Biopsie d’une amygdale d’un malade de la variante de la maladie de Creutzfeldt-Jakob. © Sbrandner, cc by-sa 3.0
Tissus où le prion anormal s'accumule
Les signes moléculaires diffèrent aussi suivant les maladies. « Alors que le prion est essentiellement retrouvé dans le système nerveux centralsystème nerveux central pour la majorité des formes des maladies à prions, pour la forme variante (vache folle) on le retrouve aussi dans les organes lymphoïdeslymphoïdes (ganglionganglion, raterate, appendice...). Pour cette forme particulière, une biopsiebiopsie des amygdales pharyngées constitue donc un argument diagnostique. C'est en raison de la présence de PrP anormaux en périphérie que des cas de contaminationcontamination suite à des transfusions sanguines ont été observés (4 en Grande-Bretagne). », explique Jean-Philippe Brandel.

par Claire Peltier, Futura
Bons plans

Conso
Guides fitness et bien-être
Soldes : le tapis de course Citysports ZX1 est à portée de main sur Cdiscount

Conso
Guides fitness et bien-être
Quels sont les meilleurs tapis de marche à moins de 200 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 600 € ?

Conso
Guides fitness et bien-être
Fitness à domicile : -240 € sur ce rameur pliable BIGZZIA dont le prix fait sensation !

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 500 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 400 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 300 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 200 € ?

Conso
Guides fitness et bien-être







, Wikimedia Commons, DP")
