










au sommaire
Les différentes formes de la maladie de Creutzfeldt-Jakobmaladie de Creutzfeldt-Jakob (maladie à prionprion) varient en fonction de leur mode d'apparition : acquise, sporadique ou génétiquegénétique.


La maladie de la vache folle a provoqué la maladie de Creutzfeldt-Jakob. © Pexels, CCO
Maladie à prion : la forme acquise
Les formes de la maladie de Creutzfeldt-Jakob d'origine infectieuse sont notamment observées chez les enfants traités par l'hormonehormone de croissance, mais d'autres contaminationscontaminations ont eu lieu suite à des greffesgreffes. Des greffes de dure-mèredure-mère, une membrane rigide qui protège le cerveaucerveau, ont mené à la contamination de nombreux japonais dans les années 1980, mais aussi de 13 personnes en France. « Ces dure-mères avaient été prélevées sur des cadavres », comme les hypophyseshypophyses dans les cas de l'hormone de croissance. On note également quelques cas de contamination après des greffes de cornée. Des actes chirurgicaux effectués avec du matériel insuffisamment stérilisé pour éliminer le prion ont également contaminé de rares patients.
 a provoqué des cas de variante de la maladie de Creutzfeldt-Jakob chez des consommateurs de viande bovine. © DR")
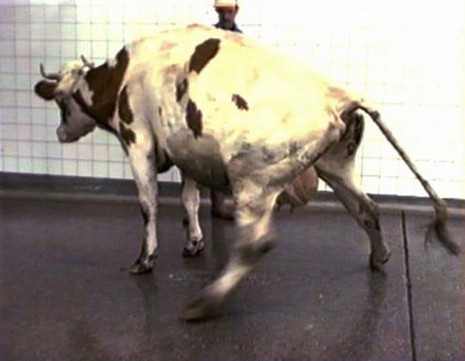
La maladie de la vache folle ou encéphalopathie spongiforme bovine (ESB) a provoqué des cas de variante de la maladie de Creutzfeldt-Jakob chez des consommateurs de viande bovine. © DR
Une autre forme infectieuse dont on a beaucoup parlé est la variante de la maladie de Creutzfeldt-Jakob (VMCJ), liée à la vache follevache folle. Les personnes se sont contaminées « par l'ingestioningestion de dérivés bovins, probablement pas de viande au sens du muscle, mais probablement par la cervelle de bovin. » explique Jean-Philippe Brandel. Au total, il y eu un peu plus de 170 cas en Grande-Bretagne, 25 cas en France et quelques cas dans d'autres pays.
Maladie de Creutzfeldt-Jakob : la forme sporadique
La forme sporadique de la maladie de Creutzfeldt-Jakob représente 80 % des cas de maladies à prions. Elle survient spontanément sans que l'on en connaisse réellement la cause. « On ne sait d'ailleurs toujours pas si les gens se sont contaminés très longtemps auparavant ou si la protéineprotéine PrPc normale cellulaire est devenue spontanément anormale. », explique Jean-Philippe Brandel. On suppose néanmoins qu'elle survient spontanément car on ne recense aucun risque de contamination dans les antécédents des patients.
« Cette forme sporadique touche surtout les personnes âgées autour de 60 à 70 ans, comme les autres maladies neurodégénérativesmaladies neurodégénératives (Alzheimer, Parkinson). L'incidenceincidence de cette forme de maladie de Creutzfeldt-Jakob est d'environ 1 à 2 cas par million d'habitant, elle existe partout et on sait qu'elle est transmissible. C'est cette forme qui a été décrite au départ par Creutzfeldt en 1920 puis par Jakob en 1921.»
La forme génétique
La forme génétique représente 10 % des cas de maladie de Creutzfeldt-Jakob. Les personnes atteintes possèdent une version anormale du gène de la protéine cellulaire PrPc, codé par le chromosomechromosome 20. La protéine prion synthétisée porteporte alors des anomaliesanomalies et va avoir plus de risques de se conformer de manière anormale et de provoquer la maladie. « Le gènegène anormal se transmet de génération en génération selon un mode autosomiqueautosomique dominant, c'est-à-dire qu'on a un risque sur deux d'être porteur du gène anormal si un des deux parents est porteur de cet allèleallèle anormal ».
Les personnes qui naissent avec une protéine mutée ont donc plus de risques de développer une maladie à prion que quelqu'un qui n'a pas de protéine anormale au départ. Pendant longtemps ils vivent très bien, mais la maladie se déclare en moyenne vers 40-50 ans, c'est-à-dire généralement plus précocement que la forme sporadique. En France, il y a 10 à 15 cas de cas maladies génétiquesmaladies génétiques à prion par an, c'est donc très rare.

par Claire Peltier, Futura
Bons plans

Conso
Guides fitness et bien-être
Soldes : le tapis de course Citysports ZX1 est à portée de main sur Cdiscount

Conso
Guides fitness et bien-être
Quels sont les meilleurs tapis de marche à moins de 200 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 600 € ?

Conso
Guides fitness et bien-être
Fitness à domicile : -240 € sur ce rameur pliable BIGZZIA dont le prix fait sensation !

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 500 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 400 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 300 € ?

Conso
Guides fitness et bien-être
Quelles sont les meilleures stations de musculation à moins de 200 € ?

Conso
Guides fitness et bien-être






 dans le cerveau d'un enfant atteint de creutzfeldt-jacob iatrogene
Crédit: Fournier J.G. INSERM")
 s'accumulent entre les neurones (en bleu) et sont liées à l’agrégation de protéines mal repliées. Le phénomène est tout à fait similaire dans les maladies à prions. © Juan Gaertner, Shutterstock")


