

Une nouvelle étude décrit le traitement par remplacement enzymatique d'un fœtus atteint de la maladie de Pompe infantile, une maladie de stockage lysosomal. Les bons résultats encouragent la poursuite des recherches sur les thérapies moléculaires prénatales.
au sommaire
Pour la première fois, une maladie génétique rare a été traitée avant la naissance à l'aide d'une thérapie de remplacement d'enzymesenzymes, directement dans l'utérus. La réussite est le fruit d'une collaboration entre l'université de Californie à San Francisco (où se déroule actuellement un essai clinique du traitement), l'hôpital d'Ottawa au Canada (où le fœtus a été diagnostiqué et traité) et l'université Duke. Les résultats de la nouvelle étude sont publiés dans The New England Journal of Medicine.
Une maladie de surcharge lysosomale en thérapie prénatale
La maladie de Pompe infantile qui touchait le fœtus est l'un des nombreux troubles lysosomaux rares (moins d'une naissance sur 100 000) qui peuvent endommager gravement les principaux organes avant la naissance. Il s'agit d'une maladie génétique liée à un mauvais fonctionnement de l'alpha-glucosidase acideacide, une enzyme qui décompose le glycogène en glucoseglucose. Le glycogène s'accumule alors dans l'organisme, ce qui entraîne une faiblesse musculaire progressive. Les bébés atteints par cette maladie ont un cœur hypertrophié et meurent le plus souvent à l'âge de 2 ans s'ils ne sont pas traités.
La maladie est appelée « de surcharge lysosomale » car l'enzyme défaillante est localisée au niveau du lysosomelysosome. « Les patients atteints de maladies de stockage lysosomal à début précoce sont des candidats idéaux pour une thérapie prénatale, car les dommages aux organes commencent in utero », écrivent les chercheurs. Le dépistagedépistage très précoce a permis la mise en place dans l'utérus d'un traitement enzymatiqueenzymatique substitutif (TES) avec des enzymes recombinantes, sur la base de précédents résultats réussis chez la souris.
 et de la patiente actuelle (colonne de droite) ; le panneau B montre une coloration PAS (sans digestion par la diastase) des villosités placentaires. Le traitement in utero a empêché l'accumulation pathologique de glycogène. © Cohen, Chakraborty, Fung-Kee-Fung <em>et al. The NEJM</em> (2022)")
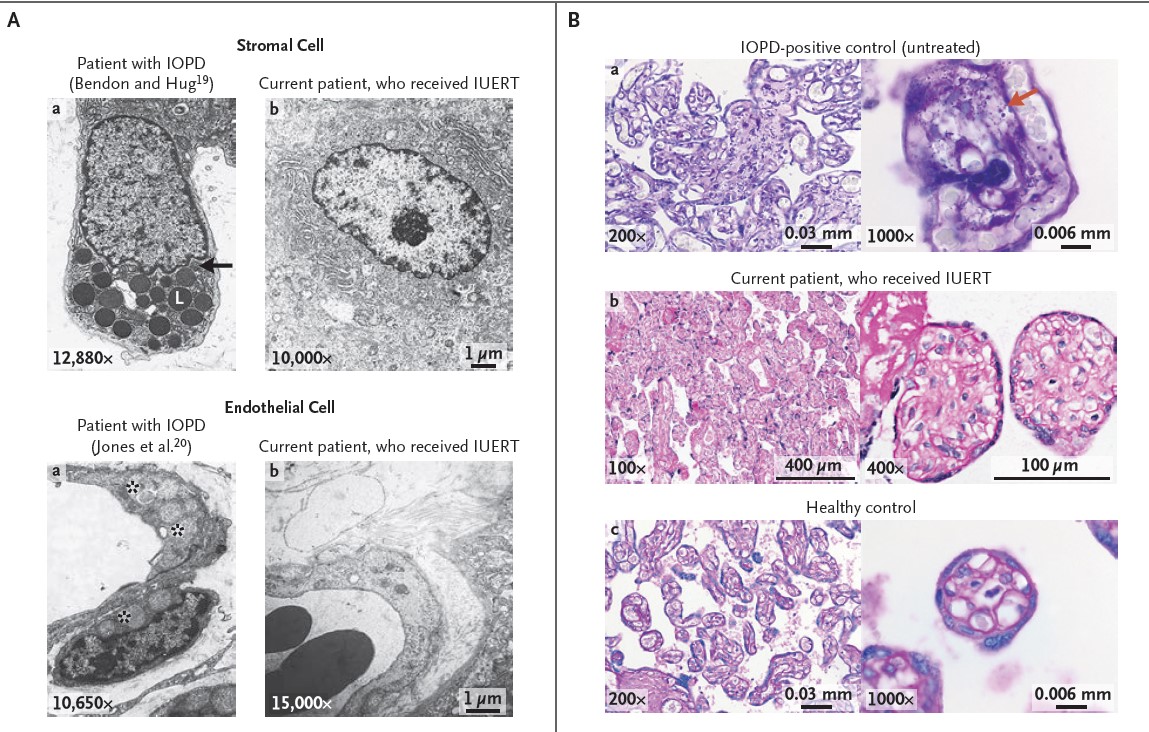
Un traitement in utero prometteur
Les chercheurs ont obtenu l'approbation accélérée de la Food and Drug AdministrationFood and Drug Administration pour un essai clinique sur dix humains, puis ont testé le traitement sur un patient éligible. Les parents d'Ayla ont découvert que leur enfant (encore un fœtus à ce moment-là) était atteinte de la même maladie génétiquemaladie génétique que celle qui a coûté la vie à deux de leurs enfants. Les chercheurs rapportent la consanguinitéconsanguinité des parents dans leur étude.
“La patiente présente des fonctions cardiaque et motrice normales”
À l'hôpital d'Ottawa, Ayla a reçu six traitements prénataux par remplacement enzymatique. Née à terme, elle a maintenant 16 mois et suit un traitement enzymatique postnatal. En effet, les patients atteints de maladies de stockage lysosomal doivent recevoir des perfusionsperfusions intraveineuses d'enzymes toutes les quelques semaines pendant toute leur vie, même si les chercheurs misent sur les progrès de la recherche pour s'en passer. « Après avoir reçu un TES in utero et un traitement postnatal standard, la patiente présente des fonctions cardiaque et motrice normales après la naissance, des biomarqueurs de niveaux normaux, et répond aux critères de développement », écrivent les chercheurs. Ces derniers rapportent un traitement sûr pour la mère et le fœtus, ainsi qu'une efficacité dans la réduction des dépôts de glycogène placentaire. D'après eux, il s'agit d'une étape importante vers des thérapies plus curatives qui pourraient être administrées avant la naissance, comme la thérapie génique et l'édition de gènesgènes.
























